InteractionMap powered by F-SAPT: Advanced Insights into Molecular Interactions
InteractionMap powered by F-SAPT or Functional-group Symmetry-Adapted Perturbation Theory (F-SAPT) is a powerful method for computing and analyzing the interaction energy between non covalently bonded molecules. By providing detailed insights into these interactions, InteractionMap plays a crucial role in drug design and molecular engineering, helping researchers optimize molecular structures for better performance and efficacy.
.webp)
Key Features


Elevate Your Research
with InteractionMap powered by F-SAPT
Accelerate Research Timelines
Experience up to 100x faster calculations compared to legacy software, allowing you to complete complex simulations in a fraction of the time.
Gain Detailed Molecular Insights
Break down interaction energies into their fundamental components to understand the specific forces driving molecular interactions, aiding in rational molecular design.
.webp)
Improve Accuracy and Reliability
Leverage advanced GPU technology to perform high-accuracy quantum mechanical simulations, ensuring reliable results for informed decision-making.
Enhance Research Productivity
Automated structure preparation and user-friendly interfaces enable researchers to focus on innovation and discovery.
Scalability and User Experience
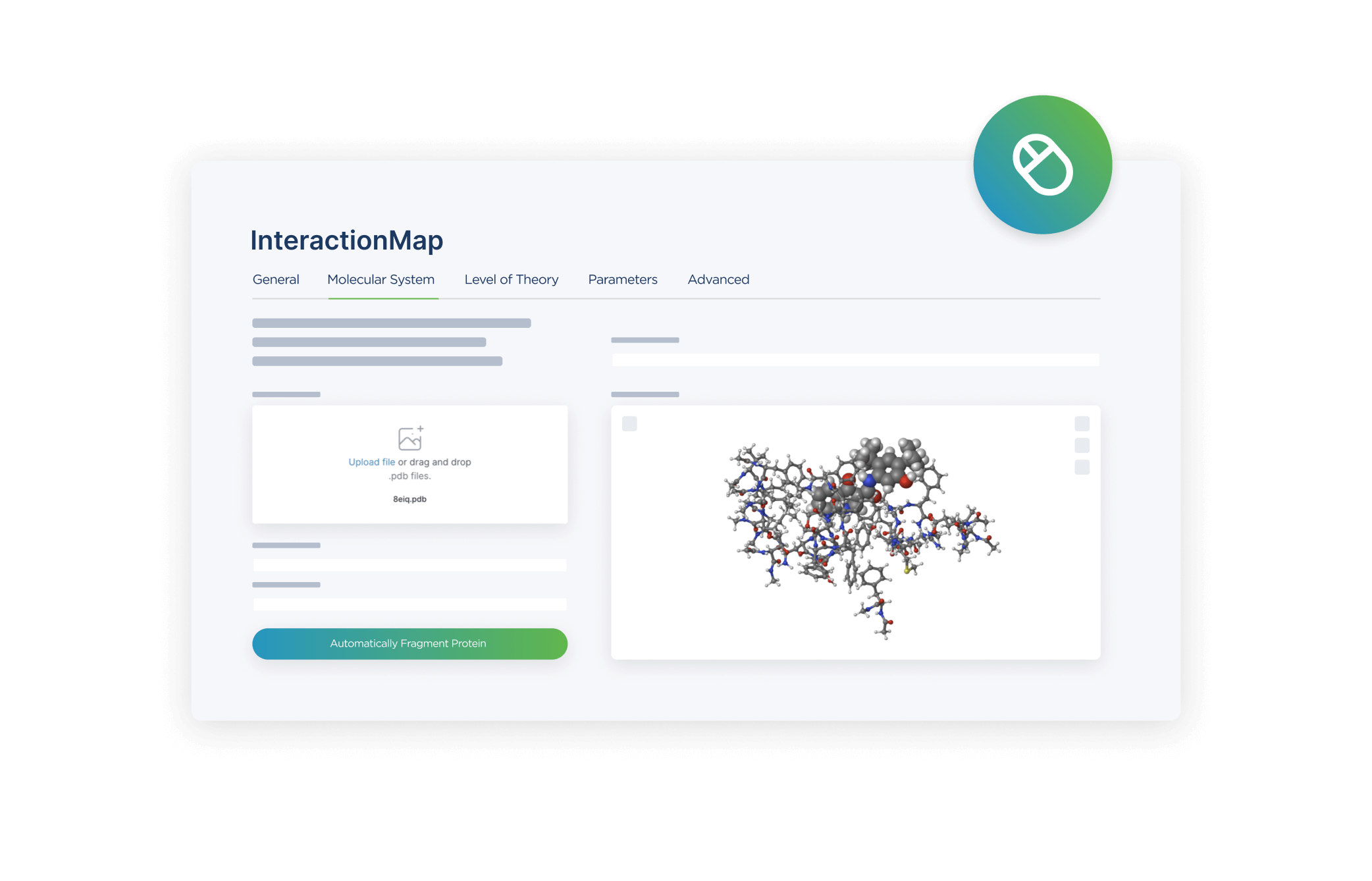
User Interface
Promethium features an intuitive graphical user interface (GUI) that simplifies the setup and execution of complex InteractionMap calculations. The platform also offers API access for advanced users and integration with external tools.
.webp)
Scalability
The platform's architecture allows for seamless scalability, leveraging AWS cloud infrastructure to handle extensive computational workloads. Batch processing capabilities enable the analysis of large datasets, making Promethium suitable for high-throughput research environments.
Frequently Asked Questions (FAQs)
Address common questions, incorporating long-tail keywords and providing clear, concise answers that add value for the reader.

InteractionMap powered by F-SAPT (Functional-group Symmetry-Adapted Perturbation Theory) is a method integrated into Promethium that computes and analyzes the interaction energy between noncovalently bonded molecules. By computing these interactions in terms of fundamental physical contributions and then decomposing each component into pairwise contributions, F-SAPT provides detailed insights into the forces driving molecular behavior. This level of analysis is essential for optimizing molecular structures in drug design and materials science, leading to more effective and efficient research outcomes.
Promethium leverages advanced GPU technology, specifically NVIDIA's A100 GPUs, to perform InteractionMap calculations with exceptional speed and efficiency. The platform's algorithms are optimized to fully utilize GPU capabilities, making complex quantum mechanical simulations significantly faster than with legacy software. This ensures that users can handle large datasets and large, complex systems.
InteractionMap powered by F-SAPT calculations on Promethium provide several detailed outputs:
- Interaction Energy: The total interaction energy of nonbonded complexes.
- Physical Contributions: Breakdown of the interaction energy into components like electrostatics, exchange-repulsion, induction, and dispersion.
- Fragment-Pair Contributions: Partitioning of interaction components into user-defined fragment pairwise contributions, offering granular insights into molecular interactions.
These outputs help researchers understand the specific forces driving molecular interactions, enabling targeted optimizations and more informed decision-making in their research.
Promethium features an intuitive graphical user interface (GUI) and API access, making it easy for both novice and advanced users to set up and execute F-SAPT calculations. The platform automates the preparation of protein-ligand complexes suitable for F-SAPT calculations. Results analysis of F-SAPT calculations on one or a series of related complexes is also automated, further streamlining the workflow. This user-friendly design ensures that sophisticated computational tools are accessible and efficient, enhancing overall research productivity.
InteractionMap powered by F-SAPT on Promethium is designed for researchers and scientists in fields such as drug discovery and materials science who need detailed and accurate insights into molecular interactions. Whether you are optimizing drug candidates or designing new materials, F-SAPT provides the advanced computational capabilities necessary to accelerate your research and achieve more effective outcomes.